
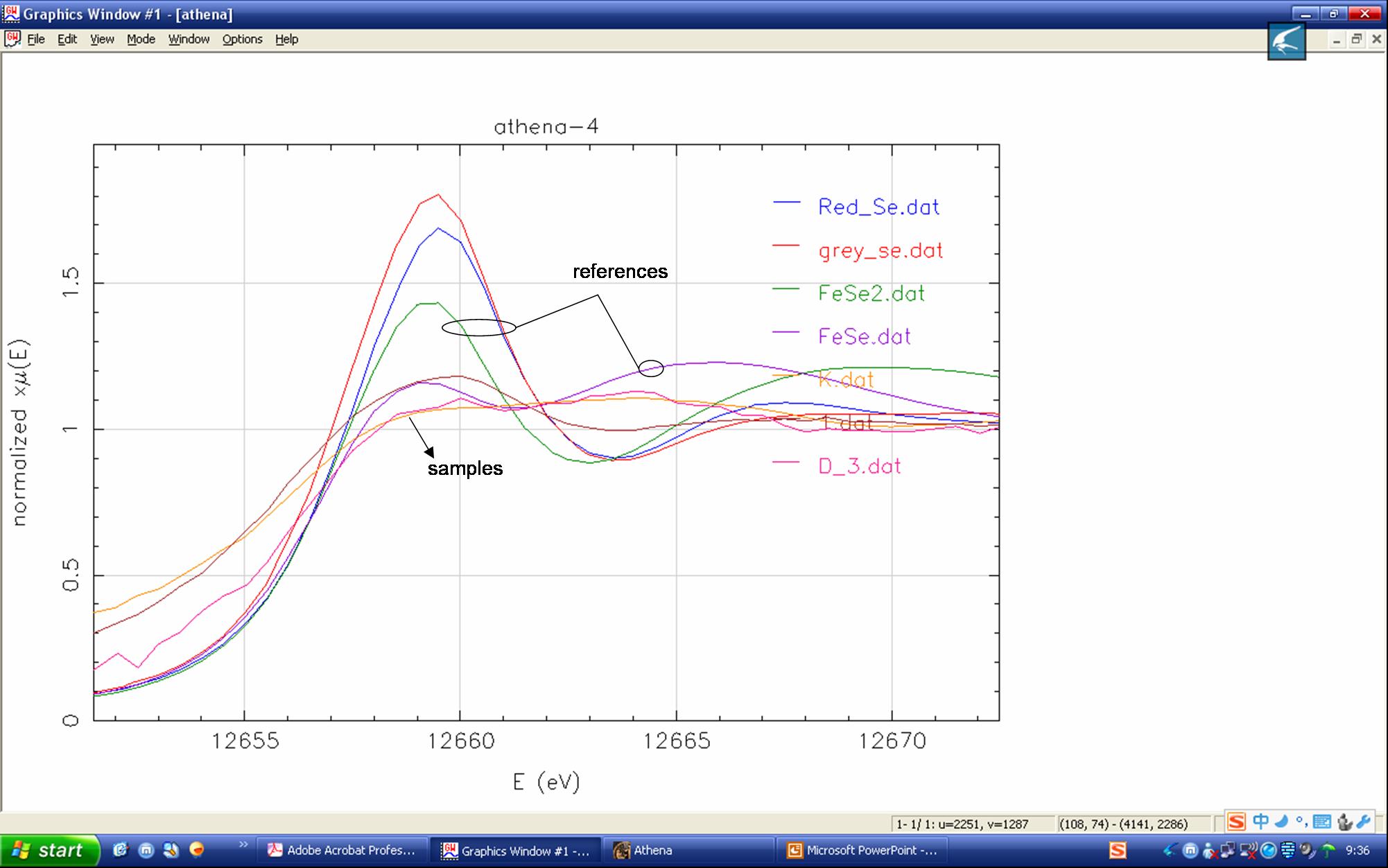
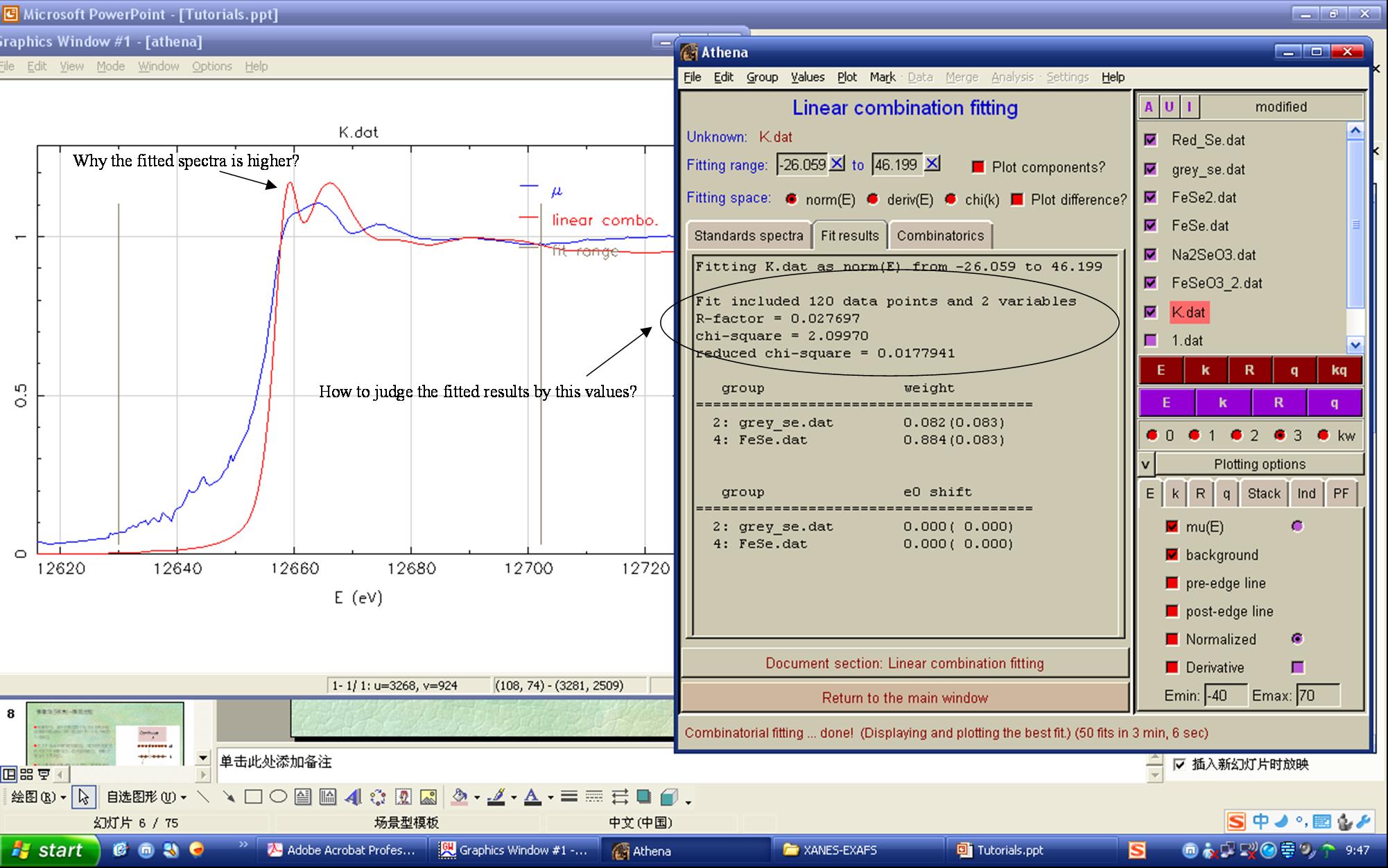
Dear, I want to to ask a question about using Linear Combination Fitting in Athena. I normalized all the references(measured in transmission mode) and samples (fuorescence mode) with Athena, and then wish to use LCF to quantify each unique component in the samples. But I noted that the normalized samples spectra had lower height at the white line peak than any references(shown pic-1.emf in annex), thus the fitted spectra were also higher than the original sample spectra (shown pic-2 and -3.emf in annex). I do not know why? I also attached all the original data(before normalization). Wish to obtain guidance from you. Thanks! Best wishes Mingliang
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

Dear Mingliang, simply because Athena is not normalising the height of the highest peak (the white line) but the EXAFS region, that is it starts some 10 eV above the edge. You choose the normalisation region in Athenas settings. You can see in your plots that it does this job quite well, the spectra oscillate around 1 (at E > E0+40 eV). Anyway, take care what you do when you try to fit a fluorescence yield XAFS spectrum with references which were measured in transmission mode. You must at least be sure that you considered the "self absorption effect" which dampens the oscillations (most notably the large oscillations in the XANES region). The importance of this effect depends on your sample's composition. Athena contains functions to correct the self absorption. In order to do this you need the elemental composition of your sample and the angles between sample, incoming beam and detector. Best regards, Edmund Welter ������ wrote:
Dear, I want to to ask a question about using Linear Combination Fitting in Athena. I normalized all the references(measured in transmission mode) and samples (fuorescence mode) with Athena, and then wish to use LCF to quantify each unique component in the samples. But I noted that the normalized samples spectra had lower height at the white line peak than any references(shown pic-1.emf in annex), thus the fitted spectra were also higher than the original sample spectra (shown pic-2 and -3.emf in annex). I do not know why? I also attached all the original data(before normalization). Wish to obtain guidance from you. Thanks! Best wishes Mingliang
------------------------------------------------------------------------ �������䣬û�������ʼ�����ѵ������䣡 http://www.yeah.net/?from=o1 ------------------------------------------------------------------------
------------------------------------------------------------------------
------------------------------------------------------------------------
------------------------------------------------------------------------
------------------------------------------------------------------------
------------------------------------------------------------------------
_______________________________________________ Ifeffit mailing list Ifeffit@millenia.cars.aps.anl.gov http://millenia.cars.aps.anl.gov/mailman/listinfo/ifeffit
Dear Edmund,
Thanks for your explain. But I am still puzzled. Following your instruction, I performed Self-Absorption for the samples, choosing XANES(Fluo) from algorithm, but I did not know the real meaning of Formula. Does it mean the species formula which I wish to detect? Or the whole composition of the sample? I used natural mineral to interact with selenite, then I wish to check the speciation of the selenium reaction product. For my case, should I fill the blank with Se or FeSe or both of them (because currently I do not know the exactly containing speciation)? For the angles, normally are they 45? Becuase I measured the samples in fluorescence mode, and didnot measure the angles between sample, incoming beam and detector. I tried to input Se in the Formula entry, and select 45 for the angles, then I got a corrected spectra which was higher than the original one. But when I using this spectra for LCF analysis, I still could not got overlapping line for the fitted spectra with the sample spectra...Wish to obtain more information from you. Thanks very much.
Best wishes
Mingliang
在2010-01-29?00:37:41,"Welter,?Edmund"?
Dear?Mingliang,
simply?because?Athena?is?not?normalising?the?height?of?the?highest?peak? (the?white?line)?but?the?EXAFS?region,?that?is?it?starts?some?10?eV? above?the?edge.?You?choose?the?normalisation?region?in?Athenas?settings.? ??You?can?see?in?your?plots?that?it?does?this?job?quite?well,?the? spectra?oscillate?around?1?(at?E?>?E0+40?eV).
Anyway,?take?care?what?you?do?when?you?try?to?fit?a?fluorescence?yield? XAFS?spectrum?with?references?which?were?measured?in?transmission?mode.? You?must?at?least?be?sure?that?you?considered?the?"self?absorption? effect"?which?dampens?the?oscillations?(most?notably?the?large? oscillations?in?the?XANES?region).?The?importance?of?this?effect?depends? on?your?sample's?composition.
Athena?contains?functions?to?correct?the?self?absorption.?In?order?to?do? this?you?need?the?elemental?composition?of?your?sample?and?the?angles? between?sample,?incoming?beam?and?detector.
Best?regards, Edmund?Welter
康明亮?wrote:
?Dear, ?I?want?to?to?ask?a?question?about?using?Linear?Combination?Fitting?in? ?Athena.?I?normalized?all?the?references(measured?in?transmission?mode)? ?and?samples?(fuorescence?mode)?with?Athena,?and?then?wish?to?use?LCF?to? ?quantify?each?unique?component?in?the?samples.?But?I?noted?that?the? ?normalized?samples?spectra?had?lower?height?at?the?white?line?peak?than? ?any?references(shown?pic-1.emf?in?annex),?thus?the?fitted?spectra? ?were?also?higher?than?the?original?sample?spectra?(shown?pic-2?and? ?-3.emf?in?annex).?I?do?not?know?why??I?also?attached?all?the?original? ?data(before?normalization).?Wish?to?obtain?guidance?from?you.?Thanks! ?Best?wishes ?Mingliang ? ? ?------------------------------------------------------------------------ ?网易邮箱,没有垃圾邮件的免费电子邮箱!?http://www.yeah.net/?from=o1 ?------------------------------------------------------------------------ ? ? ?------------------------------------------------------------------------ ? ? ?------------------------------------------------------------------------ ? ? ?------------------------------------------------------------------------ ? ? ?------------------------------------------------------------------------ ? ? ?------------------------------------------------------------------------ ? ?_______________________________________________ ?Ifeffit?mailing?list ?Ifeffit@millenia.cars.aps.anl.gov ?http://millenia.cars.aps.anl.gov/mailman/listinfo/ifeffit
Thanks for your explain. But I am still puzzled. Following your instruction, I performed *Self-Absorption *for the samples, choosing *XANES(Fluo) *from *algorithm*, but I did not know the real meaning of *Formula*. Does it mean the species formula which I wish to detect? Or the whole composition of the sample? The composition of the whole sample. You do not need to know the species, you "only" need to know thew elemental composition of your sample. A hairy task if you don't digest your sample completely and put it into an ICP-MS or anything like that. I don't know which precision is necessary to yield usable results. Maybe someone else tested or calculated it. In principle the matrix produces just a smooth background absorption and knowing the composition it is easy to calculate the amount of this absorption using tabulated scattering factors. But it should also be
I used natural mineral to interact with selenite, then I wish to check the speciation of the selenium reaction product. For my case, should I fill the blank with Se or FeSe or both of them (because currently I do not know the exactly containing speciation)? As said before, the "chemical Formula" of the entire sample. "Chemical
Dear Mingliang, possible to determine this background experimentally, for instance extrapolating the background before the edge. But for this you would need a blank spectrum to "calibrate" the mued value. If you follow the link to the "document section" in Athena a very instructive webpage will open and among other things you will find citations of the original papers underlying the four different algorithms used for the self absorption correction. formula" here does not mean you have any kind of chemical bond, it just means molar ratio.
For the angles, normally are they 45? Becuase I measured the samples in fluorescence mode, and didnot measure the angles between sample, incoming beam and detector. I tried to input *Se *in the *Formula* entry, and select 45 for the angles, then I got a corrected spectra which was higher than the original one.
But when I using this spectra for LCF analysis, I still could not got overlapping line for the fitted spectra with the sample spectra... Using pure Se in the self absorption correction is definitely wrong, it overestimates the effect dramatically. Actually the effect more or less disappears for diluted samples. But even if the spectra were properly corrected I can not see any reason why the used set of reference spectra MUST fit the spectrum of your sample. There are many possible reasons for this, the most likely is that you are missing one or more important reference spectrum (Or you are not using enough references/free
45 degrees is the usually used geometry. However, the program asks for the geometry which you used not for the usually used geometry! If it was 45 degrees than its fine. Obviously the algorithm does what it is supposed to do, it amplifies the oscillations. But this does not mean that the result is correct! It will only be "correct" if the input parameters were correct. parameters in your fit...;-) Best regards, Edmund Welter

Mingliang,
For more information on self-absorption, read the links found here:
http://www.xafs.org/Experiment/OverAbsorption
This includes a link to a discussion on the mail list about self-absorption
and how to understand it.
-Richard
2010/1/29 Welter, Edmund
Dear Mingliang,
Thanks for your explain. But I am still puzzled. Following your
instruction, I performed *Self-Absorption *for the samples, choosing *XANES(Fluo) *from *algorithm*, but I did not know the real meaning of *Formula*. Does it mean the species formula which I wish to detect? Or the whole composition of the sample?
The composition of the whole sample. You do not need to know the species, you "only" need to know thew elemental composition of your sample. A hairy task if you don't digest your sample completely and put it into an ICP-MS or anything like that. I don't know which precision is necessary to yield usable results. Maybe someone else tested or calculated it. In principle the matrix produces just a smooth background absorption and knowing the composition it is easy to calculate the amount of this absorption using tabulated scattering factors. But it should also be possible to determine this background experimentally, for instance extrapolating the background before the edge. But for this you would need a blank spectrum to "calibrate" the mued value. If you follow the link to the "document section" in Athena a very instructive webpage will open and among other things you will find citations of the original papers underlying the four different algorithms used for the self absorption correction.
I used natural mineral to interact with selenite, then I wish to check the
speciation of the selenium reaction product. For my case, should I fill the blank with Se or FeSe or both of them (because currently I do not know the exactly containing speciation)?
As said before, the "chemical Formula" of the entire sample. "Chemical formula" here does not mean you have any kind of chemical bond, it just means molar ratio.
For the angles, normally are they 45? Becuase I measured the samples in
fluorescence mode, and didnot measure the angles between sample, incoming beam and detector. I tried to input *Se *in the *Formula* entry, and select 45 for the angles, then I got a corrected spectra which was higher than the original one.
45 degrees is the usually used geometry. However, the program asks for the geometry which you used not for the usually used geometry! If it was 45 degrees than its fine. Obviously the algorithm does what it is supposed to do, it amplifies the oscillations. But this does not mean that the result is correct! It will only be "correct" if the input parameters were correct.
But when I using this spectra for LCF analysis, I still could not got
overlapping line for the fitted spectra with the sample spectra...
Using pure Se in the self absorption correction is definitely wrong, it overestimates the effect dramatically. Actually the effect more or less disappears for diluted samples. But even if the spectra were properly corrected I can not see any reason why the used set of reference spectra MUST fit the spectrum of your sample. There are many possible reasons for this, the most likely is that you are missing one or more important reference spectrum (Or you are not using enough references/free parameters in your fit...;-)
Best regards, Edmund Welter
_______________________________________________ Ifeffit mailing list Ifeffit@millenia.cars.aps.anl.gov http://millenia.cars.aps.anl.gov/mailman/listinfo/ifeffit
Dear, I want to ask why the exported Chi(K) file from Artermis can not repeat the figure like in Graphic window #1 - [Athena]. I planed to output the fitted data from Artemis and plot in Origin 8.0 or Excel ect, but the figure based on the output data seems strange for Chi(K) file. For Chi(R), it was ok, the shape plotted in Origin 8.0 was the same like in Graphic window #1 - [Athena]. I do not know why?In addition, I want to ask a question about Artermis fitting. I used 5 paths for fitting, and it seemed a good fitting from the figure in Graphic window #1 - [Athena]. But the results showed that only path 1 had good values of N, R-factor, Chi-square, amp and sigma^2. If I choose only path 1 for fitting, the figure in Graphic window #1 - [Athena] showed only the highest peak fitted perfectly. Thus, I am not sure how many path that I should choose for fitting(I know only path 1 is available). The problem is, how can I export the fitted results, with or without ohter paths? Tha! nks!Best wishesMingliang

On Wednesday 03 February 2010 12:06:15 pm 康明亮 wrote:
Dear, I want to ask why the exported Chi(K) file from Artermis can not repeat the figure like in Graphic window #1 - [Athena]. I planed to output the fitted data from Artemis and plot in Origin 8.0 or Excel ect, but the figure based on the output data seems strange for Chi(K) file. For Chi(R), it was ok, the shape plotted in Origin 8.0 was the same like in Graphic window #1 - [Athena]. I do not know why?In addition, I want to ask a question about Artermis fitting. I used 5 paths for fitting, and it seemed a good fitting from the figure in Graphic window #1 - [Athena]. But the results showed that only path 1 had good values of N, R-factor, Chi-square, amp and sigma^2. If I choose only path 1 for fitting, the figure in Graphic window #1 - [Athena] showed only the highest peak fitted perfectly. Thus, I am not sure how many path that I should choose for fitting(I know only path 1 is available). The problem is, how can I export the fitted results, with or without ohter paths? Tha! nks!Best wishes
Mingliang Rather than repeat what I said before, I will give you a link and let you re-read it: http://millenia.cars.aps.anl.gov/pipermail/ifeffit/2010-January/009273.html B PS. One more hint: asking clear, easy-to-understand is one of the good reasons that Athena and Artemis give you the option of saving project files. -- Bruce Ravel ------------------------------------ bravel@bnl.gov National Institute of Standards and Technology Synchrotron Methods Group at NSLS --- Beamlines U7A, X24A, X23A2 Building 535A Upton NY, 11973 My homepage: http://xafs.org/BruceRavel EXAFS software: http://cars9.uchicago.edu/~ravel/software/exafs/

Dear Mingliang,
I have a book chapter that details how Athena normalizes data and LCF.
I'll send it to you if you want.
Just email me directly at dr.sdkelly@gmail.com. It is about 8 Meg so
make sure you have room in your mail box for it.
Cheers,
Shelly
2010/1/28 康明亮
Dear, I want to to ask a question about using Linear Combination Fitting in Athena. I normalized all the references(measured in transmission mode) and samples (fuorescence mode) with Athena, and then wish to use LCF to quantify each unique component in the samples. But I noted that the normalized samples spectra had lower height at the white line peak than any references(shown pic-1.emf in annex), thus the fitted spectra were also higher than the original sample spectra (shown pic-2 and -3.emf in annex). I do not know why? I also attached all the original data(before normalization). Wish to obtain guidance from you. Thanks! Best wishes Mingliang
________________________________ 网易邮箱,没有垃圾邮件的免费电子邮箱! _______________________________________________ Ifeffit mailing list Ifeffit@millenia.cars.aps.anl.gov http://millenia.cars.aps.anl.gov/mailman/listinfo/ifeffit
participants (5)
-
 Bruce Ravel
Bruce Ravel -
 Richard Mayes
Richard Mayes -
 Shelly Kelly
Shelly Kelly -
 Welter, Edmund
Welter, Edmund -
 康明亮
康明亮