Attempted XANES calc using FEFF8.1 - what's wrong?

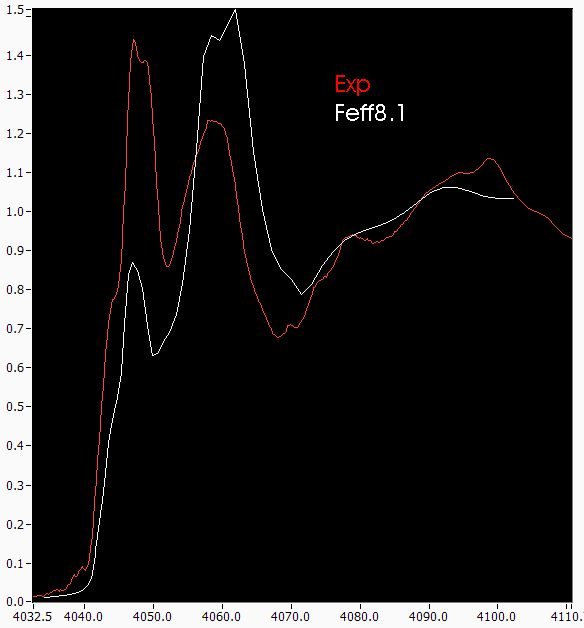
I tried the XANES calculation on FEFF8.10 with calcite at the Ca K-edge as the object. I had just taken some data on this compound, and I wanted to see how it compares. The attached shows that it doesn't compare all that well. I've also attached the feff.inp file I used, which was made by ATOMS using coordinates from the atoms.inp database. I used default parameters and read the result from the xmu.dat file. The energy cal on the experimental data was done using the Sb L3-edge assumed at 4132.2eV, since 3um foils of Ca metal are a little hard to handle :-). Next stop, I suppose, is FDMNES, which I recently downloaded. mam
{kind=link}

Hi Marcus, In general I would use slightly larger basis set than you do, i.e.. use lmax =2 for O and C, also increase the cluster size. If you look at the pDOS, you will see pretty good agreement between the *shape* of K-shell expt (xmu.dat cols 2 and 4) which probes l=1. The main problems, as I see it, is that the *amplitudes* of the 1st two peaks are inverted with FEFF8 (the 1st peak should be higher,2nd peak should be lower) and also that there is too much loss. Cheers, John On Fri, 11 Aug 2006, Matthew Marcus wrote:
I tried the XANES calculation on FEFF8.10 with calcite at the Ca K-edge as the object. I had just taken some data on this compound, and I wanted to see how it compares. The attached shows that it doesn't compare all that well. I've also attached the feff.inp file I used, which was made by ATOMS using coordinates from the atoms.inp database. I used default parameters and read the result from the xmu.dat file. The energy cal on the experimental data was done using the Sb L3-edge assumed at 4132.2eV, since 3um foils of Ca metal are a little hard to handle :-). Next stop, I suppose, is FDMNES, which I recently downloaded. mam

Dear All, Does someone happen to have a data set of Zr k- edge in metallic glass alloy, either with Pt or Cu? I just collected some data of Zr3Pt1 metallic glass ribbon in transmission and the spectrum looks wired...... Thanks, Ning Yang
OK, I used the default settings. I'm trying FDMNES now and it seems to do
better with default settings, but it's very slow,
even in 'Green' (multiple-scattering) mode. That's with a 6A cluster for
comparison.
I think there are more serious disagreements than just the amplitudes of two
peaks:
1. The pre-edge features are missing in FEFF (and FDMNES).
2. The big second feature that's split in experimental data isn't in
simulation.
mam
----- Original Message -----
From: "John J. Rehr"
Hi Marcus,
In general I would use slightly larger basis set than you do, i.e.. use lmax =2 for O and C, also increase the cluster size. If you look at the pDOS, you will see pretty good agreement between the *shape* of K-shell expt (xmu.dat cols 2 and 4) which probes l=1. The main problems, as I see it, is that the *amplitudes* of the 1st two peaks are inverted with FEFF8 (the 1st peak should be higher,2nd peak should be lower) and also that there is too much loss.
Cheers, John
On Fri, 11 Aug 2006, Matthew Marcus wrote:
I tried the XANES calculation on FEFF8.10 with calcite at the Ca K-edge as the object. I had just taken some data on this compound, and I wanted to see how it compares. The attached shows that it doesn't compare all that well. I've also attached the feff.inp file I used, which was made by ATOMS using coordinates from the atoms.inp database. I used default parameters and read the result from the xmu.dat file. The energy cal on the experimental data was done using the Sb L3-edge assumed at 4132.2eV, since 3um foils of Ca metal are a little hard to handle :-). Next stop, I suppose, is FDMNES, which I recently downloaded. mam
Ifeffit mailing list Ifeffit@millenia.cars.aps.anl.gov http://millenia.cars.aps.anl.gov/mailman/listinfo/ifeffit
Hi Matthew,
I think there are more serious disagreements than just the amplitudes of two peaks: 1. The pre-edge features are missing in FEFF (and FDMNES). 2. The big second feature that's split in experimental data isn't in simulation.
Actually some pre-edge features and the split in the 2nd feature are visible in the pDOS. To see the preedge in FEFF one has to shift the Fermi energy by 2 eV (Vr=2) to get the Fermi energy right. Also, one has to cut the loss, Vi = -.3 eV. Matt's remarks illustrate one of the problems with the defaults in FEFF8, namely that the plasmon-pole self-energy has too much loss in the near edge. It might be worthwhile to compare results with another exchange model, e.g. Dirac-Hara exchange. or ground state. John
With all the playing around one has to do to get a known structure to agree,
how can one fit
unknown structures? What started this whole thread was the talk I heard by
Wu Ziyu who claimed
to fit metalloprotein structures based on subtle features of the XANES,
while I can't even get the broad features to agree!
It makes me wonder how robust those results really are.
How, for instance, would I know that the Fermi energy needs to be shifted or
that the exchange model might be wrong?
Presumably, other systems will require other remedies. What would be nice
is if some expert in XANES calculation could
write a review showing the effects of, reasons for, and indications for use
of, these adjustments.
mam
----- Original Message -----
From: "John J. Rehr"
Hi Matthew,
I think there are more serious disagreements than just the amplitudes of two peaks: 1. The pre-edge features are missing in FEFF (and FDMNES). 2. The big second feature that's split in experimental data isn't in simulation.
Actually some pre-edge features and the split in the 2nd feature are visible in the pDOS. To see the preedge in FEFF one has to shift the Fermi energy by 2 eV (Vr=2) to get the Fermi energy right. Also, one has to cut the loss, Vi = -.3 eV. Matt's remarks illustrate one of the problems with the defaults in FEFF8, namely that the plasmon-pole self-energy has too much loss in the near edge. It might be worthwhile to compare results with another exchange model, e.g. Dirac-Hara exchange. or ground state.
John
_______________________________________________ Ifeffit mailing list Ifeffit@millenia.cars.aps.anl.gov http://millenia.cars.aps.anl.gov/mailman/listinfo/ifeffit
Hi Matthew,
With all the playing around one has to do to get a known structure to agree, how can one fit unknown structures?
I can't even get the broad features to agree! I don't agree. I think there is actually rough agreement e.g. with
All the structural information in XAS is in chi(k) - so it is most easily accessible using EXAFS analysis of the fine structure. Getting structural info from XANES is possible, but limited. For example, with MSXAN one can get nn distance. Preedge information is more electronic than structural. the pDOS and the XANES apart from amplitudes. The amplitudes are wrong, but the shapes locations are not far off.
How, for instance, would I know that the Fermi energy needs to be shifted or that the exchange model might be wrong? You can tell by how the edge looks and by looking at the broadening. It's easy for anyone with a little experience to see/understand.
Presumably, other systems will require other remedies. What would be nice is if some expert in XANES calculation could write a review showing the effects of, reasons for, and indications for use of, these adjustments.
Good idea. For a little discussion see for example, ``Calculation and interpretation of $K$-shell x-ray absorption near edge structure of transition metal oxides," H. Modrow, S. Bucher, J. J. Rehr and A. L. Ankudinov, Phys. Rev. B. B {\bf 67}, 035123 (2003). Cheers, J. Rehr
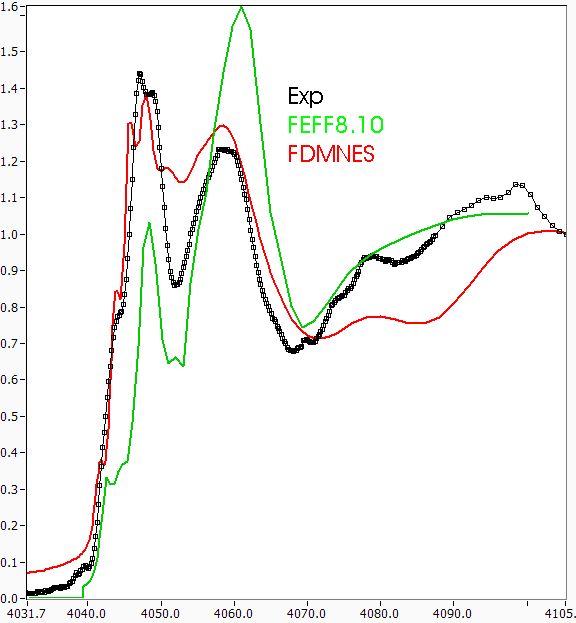
This all suggests that Ziyi Wu's claims to be able to tell the difference between 4 and 5 waters of hydration in the metal center of a protein just from fitting XANES features may be a bit overblown. I tried again with FEFF8 and still don't see the pre-edge features, nor get the ratios right. Maybe they're visible in the pDOS, but that's not what you measure. FDMNES seems to come closer to getting the ratios right, though this may be a fluke. I'll read the Modrow paper and see how they do it. mam John J. Rehr wrote:
Hi Matthew,
With all the playing around one has to do to get a known structure to agree, how can one fit unknown structures?
All the structural information in XAS is in chi(k) - so it is most easily accessible using EXAFS analysis of the fine structure. Getting structural info from XANES is possible, but limited. For example, with MSXAN one can get nn distance. Preedge information is more electronic than structural.
I can't even get the broad features to agree!
I don't agree. I think there is actually rough agreement e.g. with the pDOS and the XANES apart from amplitudes. The amplitudes are wrong, but the shapes locations are not far off.
How, for instance, would I know that the Fermi energy needs to be shifted or that the exchange model might be wrong?
You can tell by how the edge looks and by looking at the broadening. It's easy for anyone with a little experience to see/understand.
Presumably, other systems will require other remedies. What would be nice is if some expert in XANES calculation could write a review showing the effects of, reasons for, and indications for use of, these adjustments.
Good idea. For a little discussion see for example, ``Calculation and interpretation of $K$-shell x-ray absorption near edge structure of transition metal oxides," H. Modrow, S. Bucher, J. J. Rehr and A. L. Ankudinov, Phys. Rev. B. B {\bf 67}, 035123 (2003).
Cheers, J. Rehr _______________________________________________ Ifeffit mailing list Ifeffit@millenia.cars.aps.anl.gov http://millenia.cars.aps.anl.gov/mailman/listinfo/ifeffit
Here's a challenge: Suppose I have a powder of a crystal and I look at individual grains, for instance using a micro-XAS beamline. If I have good diffraction data on each grain, I could in principle use it to infer its orientation. With 6 such grains (in the general, triaxial case) I can then infer the whole absorption tensor: mu = e_transpose * Mu * e, where e is the polarization vector and Mu is the tensor which has the whole polarization dependence in it (I assume no magnetic effects and dipole approx). You can think of it as an ellipsoid which, for the general triclinic case, rotates around with respect to the crystal axes as you scan energy. One of my users has actually done this. Here's where the challenge comes in: Suppose I *don't* have any diffraction data, so I just have spectra without orientation labels. How many such spectra would I need to infer all possible data? How would I do it? Of course, it would be ambiguous with respect to rotation, so I'd have to define a set of principle axes at one energy. This job would be sort of like the kinds of reconstructions people do of TEM pix of randomly-oriented objects. Would PCA give it to me? I thought of this while taking Fe K-edge spectra of augite powder (monoclinic) and finding that the XANES spectra on four grains all looked different. mam
As John asked for, I've attached a graph showing the performance of FEFF8.10 and FDMNES in matching calcite. As Victor asked for, I've attached the calcite data, normalized. mam
{kind=link}
participants (4)
-
 John J. Rehr
John J. Rehr -
 Matthew
Matthew -
Matthew Marcus
-
 Ning Yang
Ning Yang