Variation in pre-edge intensity with replicate scans without change in EXAFS

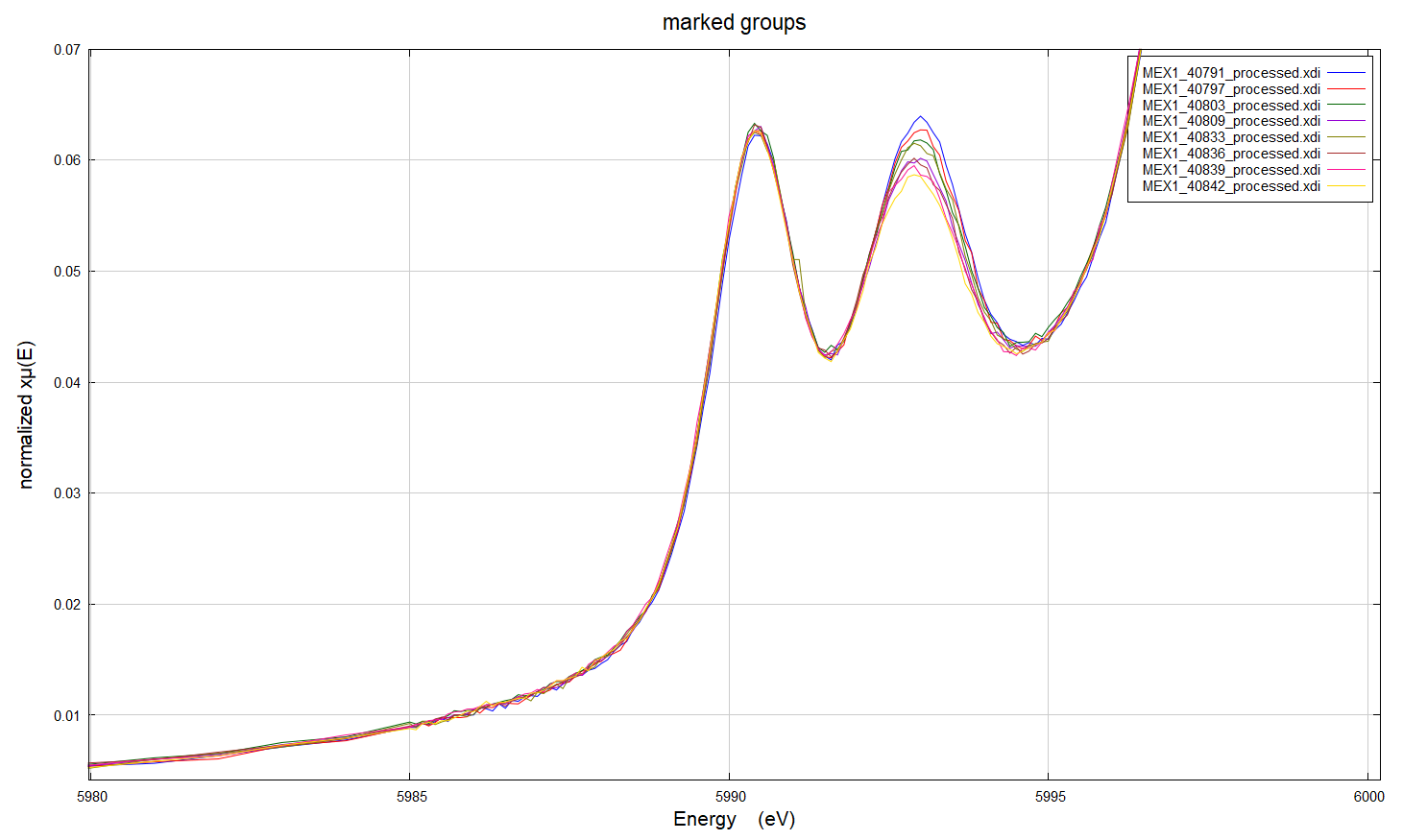
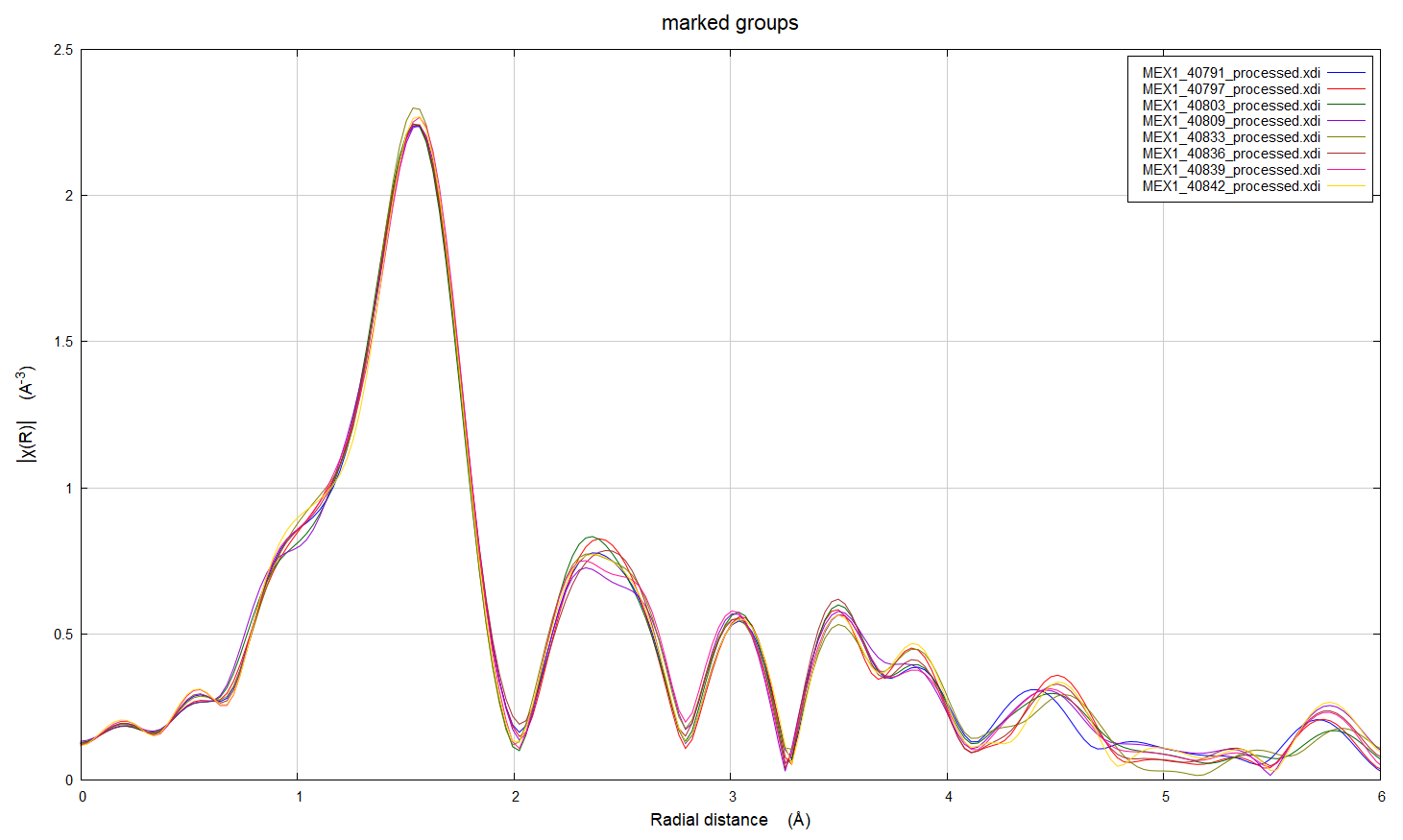
Hi everyone, I've got some XAS data at the Cr K edge for Cr-doped alumina (i.e. ruby) powder samples (Cr 0.1-1 atomic%) where we are seeing a reduction in one of the pre-edge peaks with replicate scans. There are three sets of samples; each made by a different synthetic method. We did do the repeat scans on the same spot on the samples and noted this change after the data was collected, which means that the change might be a result of beam damage. However, the results are odd because: 1. The reduction in intensity is consistent across different Cr concentrations for samples made by the same method, but is not observed for all preparation methods 2. The XAFS does not show the same systematic variation with replicate scans which suggests that this change in the pre edge is not due to a structural change 3. The edge position doesn't change with repeat scans 4. We have compositional data for these samples, and there are no significant impurities 5. I've worked up the XAFS data in Artemis and there doesn't seem to be significant differences in the structures of the samples prepared by the three different methods. For all methods, it looks like there is a second Cr atom in the second coordination shell, which is also supported by luminescence data. I've looked online and found a computational study where intensity differences in ruby samples may be related to electron spin (doi: 10.1238/Physica.Topical.115a00191, doi: 10.1039/b926499j), but I'm not sure if this is a sensible explanation - I'm still quite new to this technique. Has anyone come across anything similar, or does anyone know why we might have seen this in our samples? I've attached some images of the pre-edge and XAFS regions for the scans to illustrate what I'm asking about. Thank you, Rachel
{kind=link}
{kind=link}

Hi, Any chance of a Cr6+ contaminant? The very pronounced pre-edge feature of Cr6+ in chromate appears to align well with the second feature in Cr3+ (Cr2O3). If that contaminant is being reduced in the beam, that might explain what you are seeing. e.g. https://urldefense.us/v3/__https://www2.ung.si/*arcon/_borders/xas/xanes/xan... -R. On Wed, Aug 20, 2025 at 5:30 AM Rachel Pepper via Ifeffit < ifeffit@millenia.cars.aps.anl.gov> wrote:
Hi everyone, I’ve got some XAS data at the Cr K edge for Cr-doped alumina (i. e. ruby) powder samples (Cr 0. 1-1 atomic%) where we are seeing a reduction in one of the pre-edge peaks with replicate scans. There are three sets of samples; ZjQcmQRYFpfptBannerStart This Message Is From an External Sender This message came from outside your organization.
ZjQcmQRYFpfptBannerEnd
Hi everyone,
I’ve got some XAS data at the Cr K edge for Cr-doped alumina (i.e. ruby) powder samples (Cr 0.1-1 atomic%) where we are seeing a reduction in one of the pre-edge peaks with replicate scans. There are three sets of samples; each made by a different synthetic method.
We did do the repeat scans on the same spot on the samples and noted this change after the data was collected, which means that the change might be a result of beam damage. However, the results are odd because:
1. The reduction in intensity is consistent across different Cr concentrations for samples made by the same method, but is not observed for all preparation methods 2. The XAFS does not show the same systematic variation with replicate scans which suggests that this change in the pre edge is not due to a structural change 3. The edge position doesn’t change with repeat scans 4. We have compositional data for these samples, and there are no significant impurities 5. I’ve worked up the XAFS data in Artemis and there doesn’t seem to be significant differences in the structures of the samples prepared by the three different methods. For all methods, it looks like there is a second Cr atom in the second coordination shell, which is also supported by luminescence data.
I’ve looked online and found a computational study where intensity differences in ruby samples may be related to electron spin (doi: 10.1238/Physica.Topical.115a00191, doi: 10.1039/b926499j), but I’m not sure if this is a sensible explanation – I’m still quite new to this technique.
Has anyone come across anything similar, or does anyone know why we might have seen this in our samples?
I’ve attached some images of the pre-edge and XAFS regions for the scans to illustrate what I’m asking about.
Thank you,
Rachel ifeffit mailing list: https://millenia.cars.aps.anl.gov/mailman3/lists/ifeffit.millenia.cars.aps.a... to unsubscribe, send mail to ifeffit-leave@millenia.cars.aps.anl.gov
Hi Robert,
Thanks for responding, I don’t think that there was any formation of Cr6+ in the beam. The edge position or rest of the spectra didn’t change with repeated scans, it was just the variation in a single pre-edge peak.
Thanks again,
Rachel
From: Robert Gordon via Ifeffit

Hi Rachel,
I think you are seeing a small amount of reduction of Cr3+ to Cr2+. That looks like it could be radiation damage.
For a couple of nice references or Cr2+ and 3+ in minerals and glasses, see https://urldefense.us/v3/__https://doi.org/10.1002/cssc.201300922__;!!G_uCfs... , https://urldefense.us/v3/__https://doi.org/10.1016/j.gca.2020.09.020__;!!G_u... , and https://urldefense.us/v3/__https://doi.org/10.2138/am.2014.4646__;!!G_uCfscf... (though this last one show derivatives at the pre-edge).
One would probably expect to see only Cr3+ in alumina, and not much chance of beam damage. At least, I would expect that ;). But you would not have to reduce much of the Cr3+ to Cr2+ to see that sort of change in the pre-edge peaks. The fact that you don’t see a change in the main edge or the first shell oxygen peak are not too surprising – the overall change is small. There is a little change in the second shell, and that could mean you have some Cr not fully “in” the alumina structure, maybe interstitial or at surface or grain boundaries – which might be more susceptible to damage.
You say that impurities are low, and I would not expect alumina to have a lot of impurities, or to be hydrated. But we see beam damage with micro-XANES on hydrated minerals (and glassy phases), so I would ask whether the samples could have been hydrated or even sintered or somehow prepared with some water present?
--Matt
From: Rachel Pepper via Ifeffit
participants (3)
-
 Matthew Newville
Matthew Newville -
 Rachel Pepper
Rachel Pepper -
 Robert Gordon
Robert Gordon