distinct crystal positions for the absorber atom

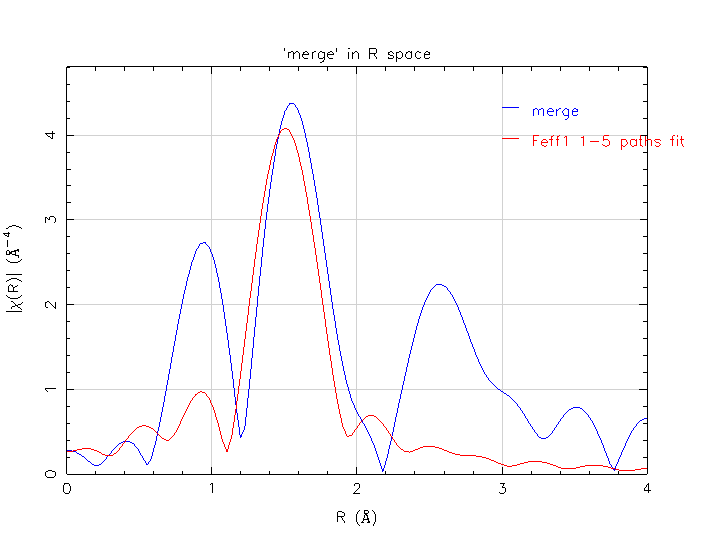
Hello everyone, I am working on fitting the first shell of a double layer vanadium oxide material. The data is V K edge data with 3 scans merged. There is two positions for vanadium in the unit structure with equal occupancy. I started modeling using equal weighting between the 2 feff calculations for each unique vanadium. Setting amp*0.5 seems straight forward. The first shell consists of oxygen coordinated and the next shell is dominated by the vanadium of the adjacent edge sharing polyhedra. Feff0 appears to give a better fit by itself with lower uncertainties than using both model in concert or Feff1 by itself. Can this problem lie in the fact that the bond lengths are quite short (1.6 A) for V-O and discerning the background and signal causes an error? I feel reasonably confident in the crystallography. I pasted the fit results below and included bmps and artemis file there. If there is something suspect please let me know or if you have a suggestion. Thank you all in advance, Christopher Patridge PhD Candidate SUNY University at Buffalo Dept of Chemistry 315-529-0501 Independent points = 7.691406250 Number of variables = 4.000000000 Chi-square = 2503.896487489 Reduced Chi-square = 678.304233648 R-factor = 0.029837426 Measurement uncertainty (k) = 0.000474272 Measurement uncertainty (R) = 0.000888467 Number of data sets = 1.000000000 Guess parameters +/- uncertainties (initial guess): amp = 0.8044660 +/- 0.1118000 (1.0000) enot = -1.9212800 +/- 1.8083430 (0.0000) ssO = 0.0001680 +/- 0.0018760 (0.0030) alpha = 0.0011620 +/- 0.0078840 (0.0000)
{kind=link}
{kind=link}
participants (1)
-
 Chris Patridge
Chris Patridge