
Dear IFEFFIT Members, I have a strange problem with FEFF calculation. I am not sure if I made some mistakes in FEFF of input file, or FEFF did not handle it well. For Oxygen paths calculated with FEFF in Artemis by 3 input files, the results for O are supposed to be close, but I got a big difference, when I transform them into r space(k 2.5~10,kweight=2 in attachment). There are three input files for FEFF: #1, 6O only on xyz axis #2, + 1P besides 6O #3, + 6P besides 6O For O, #2 and #3 are close(almost same), but #1 is much bigger. (It would make sense to me if #1 #2 are close.) Thank a lot. Best, Jack Song
{kind=link}

Jack, You have rediscovered the conventional wisdom about properly bounding a cluster for calculation with Feff. When there are not enough atoms on the periphery of the cluster to constrain the size of the muffin tins, the muffin tins tend to be much too large. As you can see this has consequence for the scattering function calculated by Feff. In your case, the muffin tins of the oxygen atoms are about 0.73 Angstroms when including even a single P atom on the periphery of the cluster. For the first calculation Rmt is 1.06 -- substantially larger. What's interesting is that a single P atom is sufficient to make the calculation of the O contribution work out reasonably well. Have you read Shelly's book chapter review of EXAFS? In her example on determining a second shell atom type, she runs Feff calculations on known crystal structures with various second shell atoms. She then takes just the kinds of paths she needs to model different contributions to the EXAFS. The reason she adopted this strategy rather than trying a small, notional cluster of the sort the you have made in your example is because constraining the muffin tins is quite important. Indeed, the potentially counter-intuitive strategy of snarfing paths from some related crystal structure works well precisely because it is a way of guaranteeing sensible muffin tin radii. HTH, B P.S. You can see the muffin tin radii by opening the .apj file as a zip file (which is what it is!) and looking at the misc.dat file in the dataX.feffY folders. On Monday, April 11, 2011 02:33:54 pm goodhei8@gmail.com wrote:
Dear IFEFFIT Members,
I have a strange problem with FEFF calculation. I am not sure if I made some mistakes in FEFF of input file, or FEFF did not handle it well.
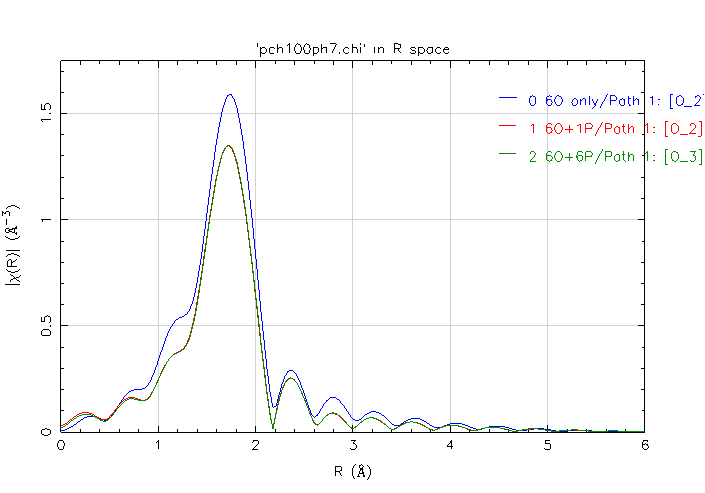
For Oxygen paths calculated with FEFF in Artemis by 3 input files, the results for O are supposed to be close, but I got a big difference, when I transform them into r space(k 2.5~10,kweight=2 in attachment).
There are three input files for FEFF: #1, 6O only on xyz axis #2, + 1P besides 6O #3, + 6P besides 6O
For O, #2 and #3 are close(almost same), but #1 is much bigger. (It would make sense to me if #1 #2 are close.)
Thank a lot.
Best,
Jack Song
-- Bruce Ravel ------------------------------------ bravel@bnl.gov National Institute of Standards and Technology Synchrotron Methods Group at NSLS --- Beamlines U7A, X24A, X23A2 Building 535A Upton NY, 11973 My homepage: http://xafs.org/BruceRavel EXAFS software: http://cars9.uchicago.edu/~ravel/software/exafs/
Dear Prof. Ravel, Thanks so much for your answer, which remove the cloud partially above me. I actually tried many models, from crystal to the opposite 6O only (different distance, second shell C,P ...). The example shown in the email are two extreme cases, which single P has contribution like 6 P. I also tried 6O+6(PO3), which is close to a crystal structure(6 O are not same distance from center, same for 6(PO3)) ---- when I decease # of PO3 from 6 to 0, FEFF calculations of O change gradually --- not like ' single P has contribution like 6 P'. It make me wonder, 1, FEFF takes Z atoms at equal distance as same. It does not care some have neighbor A, some have neighbor B. Should I worry about it? and when? 2, Does FEFF consider about valence state (like Ur 3 or Ur5 in center)? Model closer to sample, better result. Of course crystal structure is closer to real sample than models like 6O do. In practice, If I only consider single scattering and fit to n shell, can I say that it is good enough to have a model of n+1 shell? For instance I want to fit sample with O and P shells, it is good enough to have a model with Cd -O-P-O3, since O3 shell will influence P(like P atoms influence 1st shell O). Best, Jack Song On 2011-04-11 15:05, Bruce Ravel wrote:
Jack,
You have rediscovered the conventional wisdom about properly bounding a cluster for calculation with Feff. When there are not enough atoms on the periphery of the cluster to constrain the size of the muffin tins, the muffin tins tend to be much too large. As you can see this has consequence for the scattering function calculated by Feff.
In your case, the muffin tins of the oxygen atoms are about 0.73 Angstroms when including even a single P atom on the periphery of the cluster. For the first calculation Rmt is 1.06 -- substantially larger.
What's interesting is that a single P atom is sufficient to make the calculation of the O contribution work out reasonably well.
Have you read Shelly's book chapter review of EXAFS? In her example on determining a second shell atom type, she runs Feff calculations on known crystal structures with various second shell atoms. She then takes just the kinds of paths she needs to model different contributions to the EXAFS. The reason she adopted this strategy rather than trying a small, notional cluster of the sort the you have made in your example is because constraining the muffin tins is quite important. Indeed, the potentially counter-intuitive strategy of snarfing paths from some related crystal structure works well precisely because it is a way of guaranteeing sensible muffin tin radii.
HTH, B
P.S. You can see the muffin tin radii by opening the .apj file as a zip file (which is what it is!) and looking at the misc.dat file in the dataX.feffY folders.
On Monday, April 11, 2011 02:33:54 pm goodhei8@gmail.com wrote:
Dear IFEFFIT Members,
I have a strange problem with FEFF calculation. I am not sure if I made some mistakes in FEFF of input file, or FEFF did not handle it well.
For Oxygen paths calculated with FEFF in Artemis by 3 input files, the results for O are supposed to be close, but I got a big difference, when I transform them into r space(k 2.5~10,kweight=2 in attachment).
There are three input files for FEFF: #1, 6O only on xyz axis #2, + 1P besides 6O #3, + 6P besides 6O
For O, #2 and #3 are close(almost same), but #1 is much bigger. (It would make sense to me if #1 #2 are close.)
Thank a lot.
Best,
Jack Song
On Tuesday, April 12, 2011 03:24:22 am goodhei8@gmail.com wrote:
1, FEFF takes Z atoms at equal distance as same. It does not care some have neighbor A, some have neighbor B. Should I worry about it? and when?
In Feff6, two atoms that are at the same distance and have the same unique potential make the same contribution to the EXAFS. I don't quite understand what you want to worry about.
2, Does FEFF consider about valence state (like Ur 3 or Ur5 in center)?
Feff6 does not understand valence. This is one of the differences between Feff6 and Feff8. Feff8 uses a self-consistency loop to transfer charge between the atom types, resulting in something that can be related to formal valence in some situations. The follow-up question you have to ask is whether EXAFS analysis is actually sensitive to charge transfer. In the regime in which the EXAFS fit is evaluated, the photoelectron has large kinetic energy and is not particularly sensitive to the details of the potential surface. In almost all situations, Feff8 is a modest improvement at best over Feff6 for EXAFS analysis. That is why we continue to distribute and use Feff6 with Ifeffit and Artemis. Of course, charge transfer is a dramatic effect in the XANES. But you were asking a question about EXAFS analysis. B -- Bruce Ravel ------------------------------------ bravel@bnl.gov National Institute of Standards and Technology Synchrotron Methods Group at NSLS --- Beamlines U7A, X24A, X23A2 Building 535A Upton NY, 11973 My homepage: http://xafs.org/BruceRavel EXAFS software: http://cars9.uchicago.edu/~ravel/software/exafs/
Thanks a lot for the detailed answer. Best, Jack Song On 2011-04-12 09:03, Bruce Ravel wrote:
On Tuesday, April 12, 2011 03:24:22 am goodhei8@gmail.com wrote:
1, FEFF takes Z atoms at equal distance as same. It does not care some have neighbor A, some have neighbor B. Should I worry about it? and when? In Feff6, two atoms that are at the same distance and have the same unique potential make the same contribution to the EXAFS. I don't quite understand what you want to worry about.
2, Does FEFF consider about valence state (like Ur 3 or Ur5 in center)? Feff6 does not understand valence. This is one of the differences between Feff6 and Feff8. Feff8 uses a self-consistency loop to transfer charge between the atom types, resulting in something that can be related to formal valence in some situations.
The follow-up question you have to ask is whether EXAFS analysis is actually sensitive to charge transfer. In the regime in which the EXAFS fit is evaluated, the photoelectron has large kinetic energy and is not particularly sensitive to the details of the potential surface. In almost all situations, Feff8 is a modest improvement at best over Feff6 for EXAFS analysis. That is why we continue to distribute and use Feff6 with Ifeffit and Artemis.
Of course, charge transfer is a dramatic effect in the XANES. But you were asking a question about EXAFS analysis.
B
participants (2)
-
 Bruce Ravel
Bruce Ravel -
 goodhei8@gmail.com
goodhei8@gmail.com