
Hi All, I am trying to simulate the EXAFS spectrum of pure molybdenum through FEFF9 software. I got the atomic configurations of molybdenum from molecular dynamics simulations, and I'd like to import it into FEFF 9. In order to do that, I converted lammps file into cif file, and I am trying to convert cif file into feff.inp. I am using Webatoms to convert cif file into feff.inp. The default value of cluster size and longest path is 8 and 5, respectively (in angs). However, when I try to increase the cluster size and the longest path to 10, it stops working, and I got the internal error. I am working with 1024 atoms (8x8x8 cells, bcc crystal structure). Are 1024 atoms too many to deal with in webatoms? or is there other ways to convert cif file into feff.inp? I don't have any problems when I use the ideal cif file (not from molecular dynamics simulations). Sincerely, Gyuchul Park

Hi Gyuchul,
By itself, webatoms can calculate a cluster of many thousands of atoms (it
is not necessarily fast, but a cluster size of 100 Ang should work and can
give a few 100,000 atoms). Feff9, on the other hand, will have a
compiled-in maximum number of atoms in its cluster. That number could
easily be as low as 400, and you would have to rebuild Feff to make it
larger.
On Tue, Nov 19, 2019 at 8:27 PM Park Gyuchul
Hi All,
I am trying to simulate the EXAFS spectrum of pure molybdenum through FEFF9 software. I got the atomic configurations of molybdenum from molecular dynamics simulations, and I'd like to import it into FEFF 9. In order to do that, I converted lammps file into cif file, and I am trying to convert cif file into feff.inp. I am using Webatoms to convert cif file into feff.inp. The default value of cluster size and longest path is 8 and 5, respectively (in angs). However, when I try to increase the cluster size and the longest path to 10, it stops working, and I got the internal error. I am working with 1024 atoms (8x8x8 cells, bcc crystal structure). Are 1024 atoms too many to deal with in webatoms? or is there other ways to convert cif file into feff.inp? I don't have any problems when I use the ideal cif file (not from molecular dynamics simulations).
Sincerely, Gyuchul Park
_______________________________________________ Ifeffit mailing list Ifeffit@millenia.cars.aps.anl.gov http://millenia.cars.aps.anl.gov/mailman/listinfo/ifeffit Unsubscribe: http://millenia.cars.aps.anl.gov/mailman/options/ifeffit
--Matt Newville

Hi, I am wondering whether you are thinking about your problem in the most straight-forward way. Atoms is a tool to expand crystal data into a cluster. From lammps, you already have a cluster, you just don't have your cluster expressed in a form that Feff knows how to read. Unless I am significantly misunderstanding your question, I dont think Atoms is your best tool. You just need to take some (or all) of the atoms from the output cluster from your MD simulation and wrap it up in the correct dressing for Feff. Here is what I mean, explained for a much smaller problem (in terms of number of atoms): https://speakerdeck.com/bruceravel/modeling-non-crystalline-samples?slide=11 Note what I say on the next page -- the absorber DOES NOT need to be at (0,0,0) and the absorber does not need to be the first item in the list. You can just edit (or write a short script/program) your lammps output to cut out the atomic coordinates, assign unique potential indeces to all the atoms, and put the correct boilerplate at the top of the file. Voila! You have a feff input file. While it is true that you can, in principle, convert your lampps file to a CIF file, it just doesn't seem like a necessary or helpful step. I'd really recommend taking the time to understand how to move the MD output into the proper form for feff input. HTH, B On 11/19/19 9:27 PM, Park Gyuchul wrote:
Hi All,
I am trying to simulate the EXAFS spectrum of pure molybdenum through FEFF9 software. I got the atomic configurations of molybdenum from molecular dynamics simulations, and I'd like to import it into FEFF 9. In order to do that, I converted lammps file into cif file, and I am trying to convert cif file into feff.inp. I am using Webatoms to convert cif file into feff.inp. The default value of cluster size and longest path is 8 and 5, respectively (in angs). However, when I try to increase the cluster size and the longest path to 10, it stops working, and I got the internal error. I am working with 1024 atoms (8x8x8 cells, bcc crystal structure). Are 1024 atoms too many to deal with in webatoms? or is there other ways to convert cif file into feff.inp? I don't have any problems when I use the ideal cif file (not from molecular dynamics simulations).
Sincerely, Gyuchul Park
_______________________________________________ Ifeffit mailing list Ifeffit@millenia.cars.aps.anl.gov http://millenia.cars.aps.anl.gov/mailman/listinfo/ifeffit Unsubscribe: http://millenia.cars.aps.anl.gov/mailman/options/ifeffit
-- Bruce Ravel ------------------------------------ bravel@bnl.gov National Institute of Standards and Technology Synchrotron Science Group at NSLS-II Lead Beamline Scientist, 6BM (BMM) Building 743, Room 114 Upton NY, 11973 Homepage: http://bruceravel.github.io/home/ Beamline: https://www.bnl.gov/ps/beamlines/beamline.php?r=6-BM Software: https://github.com/bruceravel Demeter: http://bruceravel.github.io/demeter/
Hi Bruce Ravel,
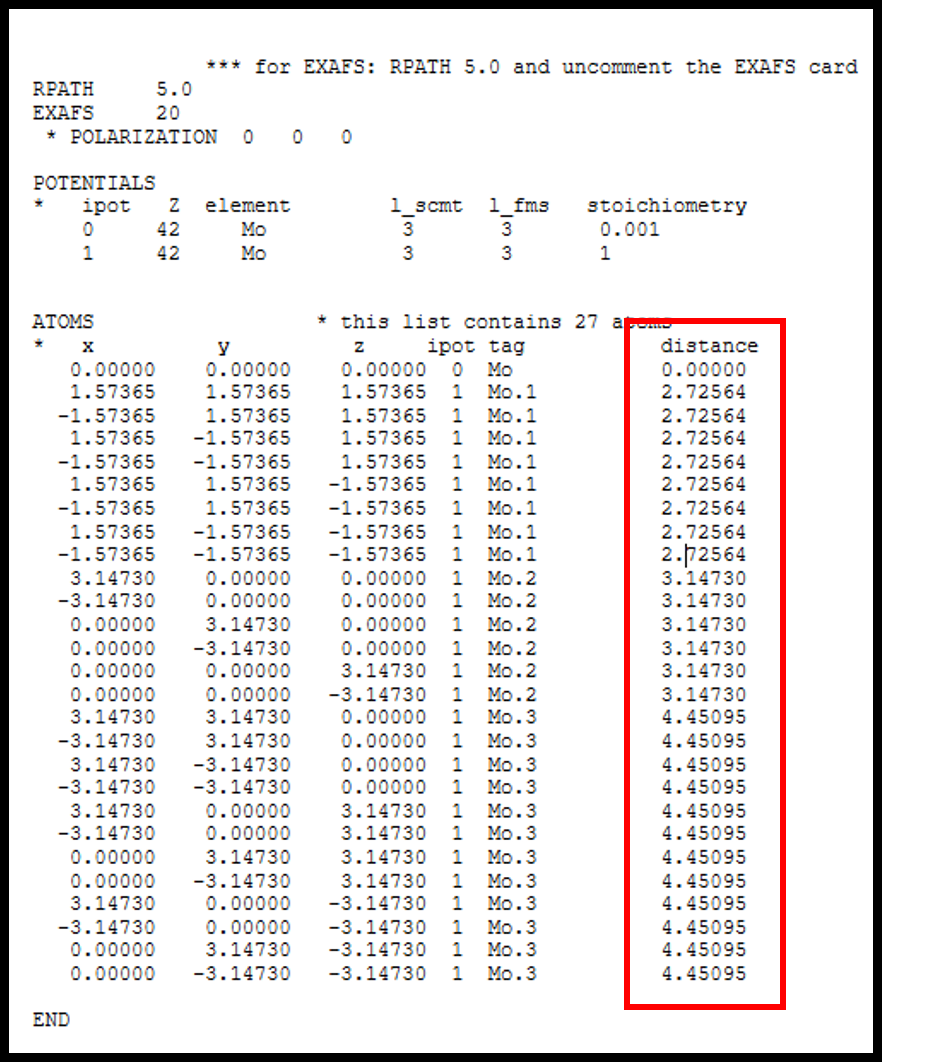
I appreciate for your precious comments. Your suggestion makes my life easier! Is "distance" column is not required information in feff.inp format? (please find the part of the feff file attached. This is what I got from WebAtoms (typical cif file is used here).
Sincerely,
Gyuchul Park
Graduate Research Assistant
School of Materials Enginnering
Purdue University
________________________________
From: Ifeffit
Hi All,
I am trying to simulate the EXAFS spectrum of pure molybdenum through FEFF9 software. I got the atomic configurations of molybdenum from molecular dynamics simulations, and I'd like to import it into FEFF 9. In order to do that, I converted lammps file into cif file, and I am trying to convert cif file into feff.inp. I am using Webatoms to convert cif file into feff.inp. The default value of cluster size and longest path is 8 and 5, respectively (in angs). However, when I try to increase the cluster size and the longest path to 10, it stops working, and I got the internal error. I am working with 1024 atoms (8x8x8 cells, bcc crystal structure). Are 1024 atoms too many to deal with in webatoms? or is there other ways to convert cif file into feff.inp? I don't have any problems when I use the ideal cif file (not from molecular dynamics simulations).
Sincerely, Gyuchul Park
_______________________________________________ Ifeffit mailing list Ifeffit@millenia.cars.aps.anl.gov http://millenia.cars.aps.anl.gov/mailman/listinfo/ifeffit Unsubscribe: http://millenia.cars.aps.anl.gov/mailman/options/ifeffit
-- Bruce Ravel ------------------------------------ bravel@bnl.gov National Institute of Standards and Technology Synchrotron Science Group at NSLS-II Lead Beamline Scientist, 6BM (BMM) Building 743, Room 114 Upton NY, 11973 Homepage: http://bruceravel.github.io/home/ Beamline: https://www.bnl.gov/ps/beamlines/beamline.php?r=6-BM Software: https://github.com/bruceravel Demeter: http://bruceravel.github.io/demeter/ _______________________________________________ Ifeffit mailing list Ifeffit@millenia.cars.aps.anl.gov http://millenia.cars.aps.anl.gov/mailman/listinfo/ifeffit Unsubscribe: http://millenia.cars.aps.anl.gov/mailman/options/ifeffit
{kind=link}
On 11/20/19 7:49 PM, Park Gyuchul wrote:
I appreciate for your precious comments. Your suggestion makes my life easier! Is "distance" column is not required information in feff.inp format? (please find the part of the feff file attached. This is what I got from WebAtoms (typical cif file is used here).
Quoting from https://bruceravel.github.io/demeter/documents/Artemis/atoms/example.html#th... "The atom list is printed in the format required by FEFF. The atom list has two comment columns. The indexed atomic symbol and radial distance are written by ATOMS for your use when reading feff.inp and are ignored by FEFF." -- Bruce Ravel ------------------------------------ bravel@bnl.gov National Institute of Standards and Technology Synchrotron Science Group at NSLS-II Lead Beamline Scientist, 6BM (BMM) Building 743, Room 114 Upton NY, 11973 Homepage: http://bruceravel.github.io/home/ Beamline: https://www.bnl.gov/ps/beamlines/beamline.php?r=6-BM Software: https://github.com/bruceravel Demeter: http://bruceravel.github.io/demeter/
participants (3)
-
 Matt Newville
Matt Newville -
 Park Gyuchul
Park Gyuchul -
 Ravel, Bruce
Ravel, Bruce