Hi Georges,
In reference to your non-critical bug. Another method to bypass this:
1) After importing CIF Cut (Right click on the line) the absorbing atom site and Paste (Right click at end of table) to the end of the atom list (ensure you re-tick CORE).
2) Run Feff and it will create the Feff input file with ipot list with the atom of interest as ipot 1 and another one as ipot [last number]
3) Delete the line with the 2nd unnecessary (final) ipot for the absorbing atom.
4) Run Feff - all should be well?
I hope this helps – credit where its due, I was shown this by another user who is on the list,
Daniel Whittaker, MChem
Nuclear FiRST DTC PhD Student
The School of Chemistry
Chemistry Building
The University of Manchester
Brunswick Street
Manchester
M13 9PL
From: Georges Siddiqi
Sent: Wednesday, October 30, 2013 8:25 PM
To: XAFS Analysis using Ifeffit
Subject: Re: [Ifeffit] Bug when adding feff calculations
thanks for noticing that!
The program I'm using (Mercury), only shows the molecule, so I guess that's why I never thought of that.
thanks again,
georges
On Wed, Oct 30, 2013 at 5:04 PM, Christopher Patridge wrote:
Hi Georges,
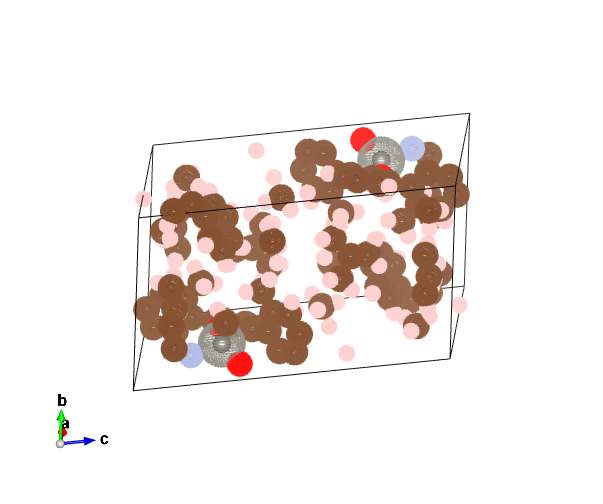
If I import the cif file into VESTA, it shows two W atoms in the unit cell explaining the addition of the second W potential in the list; attached below. The cif file only includes half of the atoms in the unit cell so it is not really a bug.
********************************************
Dr. Christopher Patridge
Assistant Professor of Chemistry
Dept of Math and Natural Science
D'youville College
320 Porter Ave., Buffalo, NY 14201
Phone: 716-829-8096
Email: patridgc@dyc.edu
On Oct 30, 2013, at 11:05 AM, Georges Siddiqi wrote:
sorry to spam, but I just received an automated message saying that the attachments were blocked because of the athena file I included:
The Copper foil.fpj has been blocked,
and Quarantine entire message has been taken on 10/30/2013 9:58:21 AM.
Message details:
Server: LUCKMAN
Sender: gsiddiqi@gmail.com;
Recipient: ifeffit@millenia.cars.aps.anl.gov;
Subject: [Ifeffit] Bug when adding feff calculations
Attachment name: Copper foil.fpj
in case the other attachments were blocked as well, I'm reattaching them.
is there a way to attach athena/artemis files without having the servers block them?
thanks,
georges
On Wed, Oct 30, 2013 at 3:55 PM, Georges Siddiqi wrote:
Hi,
I'd like to report 2 bugs that I have when dealing with Feff calculations. One is critical (program crashes), the other is more troublesome, but i've found ways around it.
OS: Windows 7, SP1
Demeter version: 0.9.18
1) Critical - Trying to add single scattering paths of arbitrary lengths. This used to work with an older version of the program, but now, no matter what I do, when I add a single scattering path with any length, the program crashes
I've attached the log file (dartemis.log)
How to replicate:
-import any file (i've attached Cu.inp for ease of comparison)
-run atoms
-run feff
-load an athena file (i've attached Cu foil.apj)
-go to "Path like" tab
-input any bond distance, and drag path over to the path list
-program should crash
This problem occurs both with cif files and inp files (and several of both file types have been tried), and (so far) any type of data set I try to import into athena. I've just chosen the attached files for their simplicity.
2) Not critical - I deal with a lot of organometallic compounds whose cif files correspond of only 1 absorbing atom surrounded by a network of non-absorbing atoms. i.e. not a regular, repeating lattice. When I import the cif file for one of these compounds (see attachment, schrock 1.cif), the Atoms page is fine, but in the Feff page, the "potentials" lists the absorbing atom as ipot 0 and 1:
POTENTIALS
* ipot Z tag
0 74 W
1 74 W
2 8 O
3 7 N
4 6 C
5 1 H
while in the atoms list, it only appears once as ipot 0. Naturally, if I just click "run Feff", the program returns an error.
My workaround to this has been to copy the atoms list into notepad, save, import into excel, subtract 1 from the ipot column (besides ipot 0), and replace the old atoms list with the corrected atoms list. then I manually change the potentials to:
POTENTIALS
* ipot Z tag
0 74 W
1 8 O
2 7 N
3 6 C
4 1 H
after this it works fine.
So this isn't a big burden, after a bit of practice I've gotten pretty fast at editing the Feff file, but if there's some easier solution it would be great.
This problem occurs with any monomeric compounds. If I run the feff on a dimeric organometallic compound, there's no problems.
Please let me know if there's any additional information I can supply that would help with this.
thanks,
Georges Siddiqi
_______________________________________________
Ifeffit mailing list
Ifeffit@millenia.cars.aps.anl.gov
http://millenia.cars.aps.anl.gov/mailman/listinfo/ifeffit
_______________________________________________
Ifeffit mailing list
Ifeffit@millenia.cars.aps.anl.gov
http://millenia.cars.aps.anl.gov/mailman/listinfo/ifeffit
--------------------------------------------------------------------------------
_______________________________________________
Ifeffit mailing list
Ifeffit@millenia.cars.aps.anl.gov
http://millenia.cars.aps.anl.gov/mailman/listinfo/ifeffit

{kind=link}