sorry for the trouble, I forgot to register this email.
-----原始邮件-----
发件人:"Ming Zhang"
发送时间:2022-01-13 14:41:04 (星期四)
收件人: ifeffit@millenia.cars.aps.anl.gov
抄送:
主题: Question--FEFF calclulated atom coordinates and labels
Dear IFEFFIT members,
I’m practicing EXAFS fitting with some standard crystal data. I GOT SOME QUESTIONS when I imported the CIF File into the FEFF calculation.
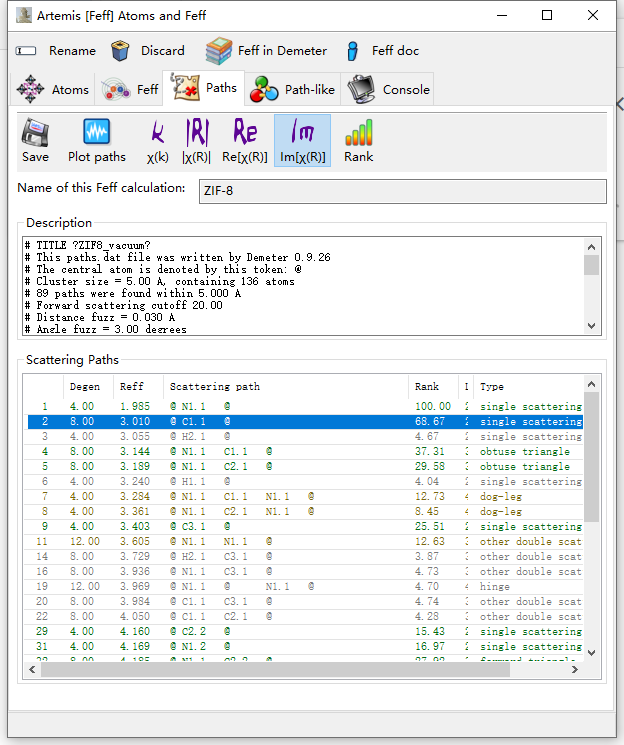
1)I’m confused with the atom label. The VESTA GUI shows that there are C1, C2, and C3 carbons in a unit cell. However, after FEFF calculation, they become C1.1 C2.1 C3.1, so do the other atoms like nitrogen and hydrogen. Since I need to set some geometry constraints, I have to figure out the nomenclature of the atom coordinates calculated by FEFF and link those atoms to real physical images.
2)I’m also confused with the calculated degeneracy, like the path of @ C1.1 @ (I have no idea about the atom C1.1 stands for). It’s a zinc absorber placed in a tetrahedron coordinated by the N atom of imidazole. Very intuitively, the degeneracy of @ N1.1 @ (row 1) is 4, and YES IT IS. But how comes the single scattering path (row 2) of the C atom next to N become 8? Also in row 5, degeneracy of @ N1.1 C2.1 @ is 8. Notably, we only got 3 C atoms in a coordinated molecule, so even treating closely located C atoms into the same one couldn’t help explain this.
FEFF calculation
VESTA GUI
The CIF file is attached. I would appreciate it very much if you could help me out.
Best regards
Ming Zhang

{kind=link}
{kind=link}