
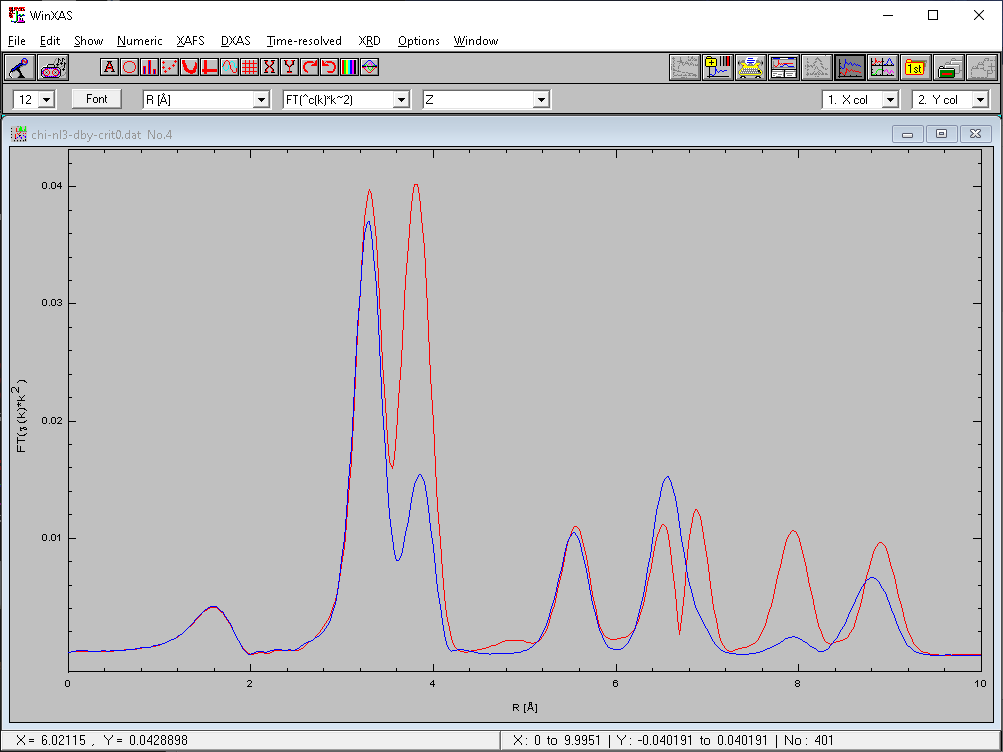
Hi Fan, I tried simulating BaSnO3 in Feff and played with the NLeg card to see the influence of multiple scattering: Blue curve is Nleg = 2 (backscattering only); red is Nleg=3 (backscatter + triangular multiple-scattering) Calcs done with DEBYE 100 307, CRITERIA 0 0, RPATH 9.5 Multiple-scattering (focused) seems to be strongly contributing both below 4Ang and below 8Ang. It seems possible that multiple scattering around the nearest neighbours may be more significant than the backscattering from 8.14Ang away. I haven't tried playing with decreasing the cluster size but keeping RPATH at 9.5Ang to see if NN MS is the culprit, but this may give you some ideas of what to check. cheers, -R. On 2024-07-05 1:45 a.m., FAN Dongxiao wrote:
Dear All, I met a problem about the misfit of EXAFS at high-range (around 8 angstrom). My target is the B site EXAFS of perovskite structure (BaSnO3, a=b=c=4. 105 angstrom) I want to study the displacement correlation between the Sn and the ZjQcmQRYFpfptBannerStart This Message Is From an External Sender This message came from outside your organization. ZjQcmQRYFpfptBannerEnd
Dear All,
I met a problem about the misfit of EXAFS at high-range (around 8 angstrom).
My target is the B site EXAFS of perovskite structure (BaSnO3, a=b=c=4.105 angstrom)
I want to study the displacement correlation between the Sn and the neighboring Sn atoms.
For the first, second, and third Sn neighboring shells, the fitting is good, but the Sn-Sn4 is bad in both amplitude and phase.
I try to include the third cumulant, not become better.
Can anybody tell me what’s the problem?
I suspect the high frequency background signal of k space to produce non-structure contribution in the FT spectra.
But the data is very clean, I think this should not be the problem.
Best,
Fan
{kind=link}