
Hi,
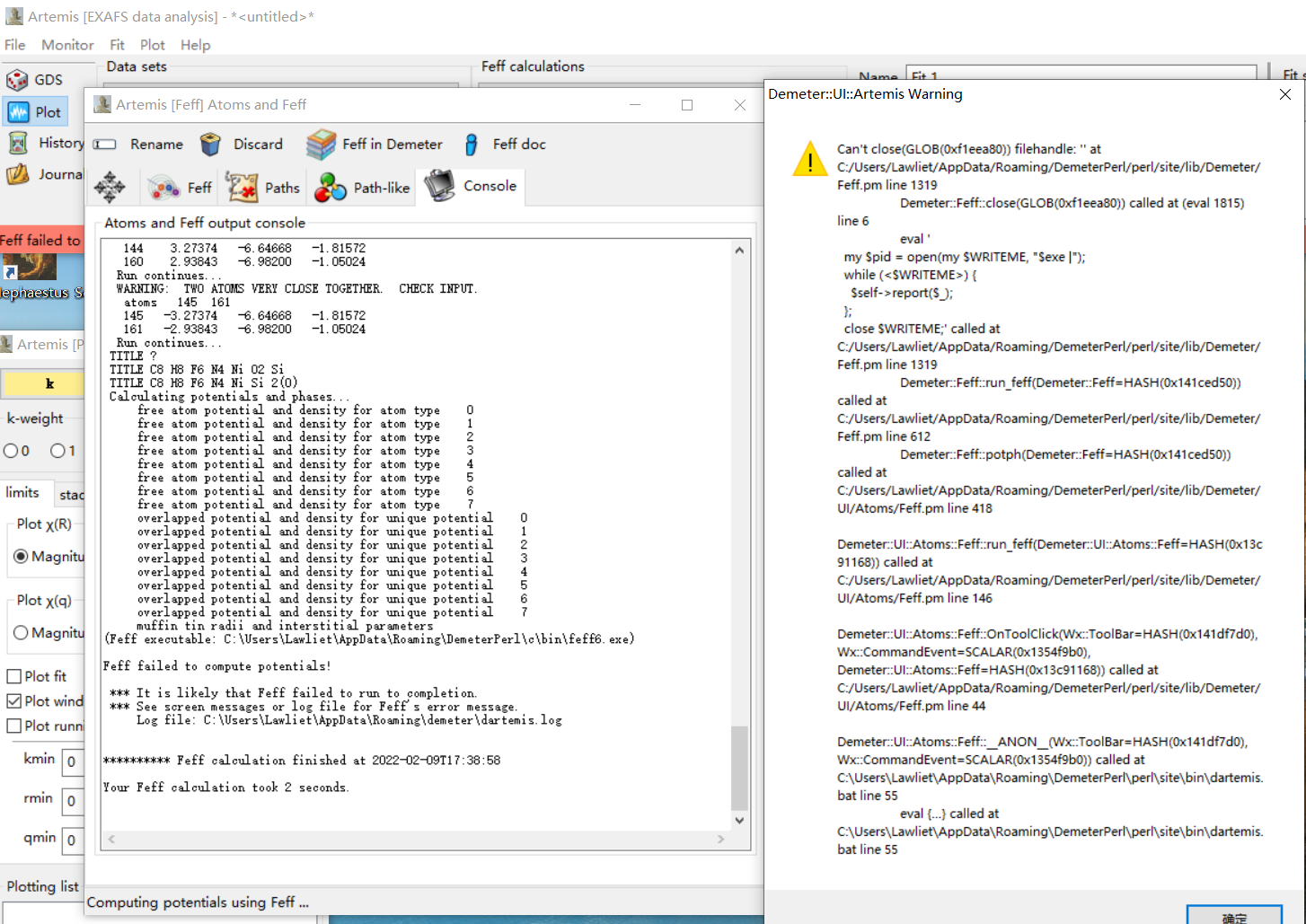
I suspect that the calculation would work much better if you removed the H
atoms. When trying to use this CIF file in Larch, I noticed 3 challenges:
a) the label of
_chemical_formula_moiety ' C8 H8 F6 N4 Ni Si, 2(O) '
doesn't parse correctly in Larch. Apparently, it does in Artemis. I
changed it to ' C8 H8 F6 N4 Ni Si O2 '.
b) the hydrogen atoms cause overlap problems. I removed these from the
`atoms` loop. This is not an uncommon problem.
c) the unit cell is large enough that one could run into a problem of not
having an atom of the same species as the central atom
(I guess Ni is what you're looking at). Artemis might not require this,
but I found that the cluster had to be at least 7.02 Ang to
make sure that a Ni scatterer was in the cluster.
Hope this helps,
On Wed, Feb 9, 2022 at 6:40 AM Xinyu Luo
Dear ifeffit memebers,
I received an Artemis warning after importing a CIF file into FEFF calculation as usual, which said, “Can’t close GLOB(…) filehandle at …” I first assumed it might be caused by some system errors. So I tried this CIF file on another PC and received a similar warning---can’t close a GLOB() filehandle. Also, I tried FEFF8 using Webatoms, but this still didn't work... Is that due to the CIF file itself or the software bug on my PC? I never met this situation before because it's always been very smooth to import a CIF file and generate atomic coordinates and paths before fitting... I don’t know how to handle this.
The CIF file is attached. I appreciate any suggestions that might be helpful. Thank you for your time!
Xinyu Luo _______________________________________________ Ifeffit mailing list Ifeffit@millenia.cars.aps.anl.gov http://millenia.cars.aps.anl.gov/mailman/listinfo/ifeffit Unsubscribe: http://millenia.cars.aps.anl.gov/mailman/options/ifeffit
-- --Matt Newville <newville at cars.uchicago.edu> 630-327-7411
{kind=link}