
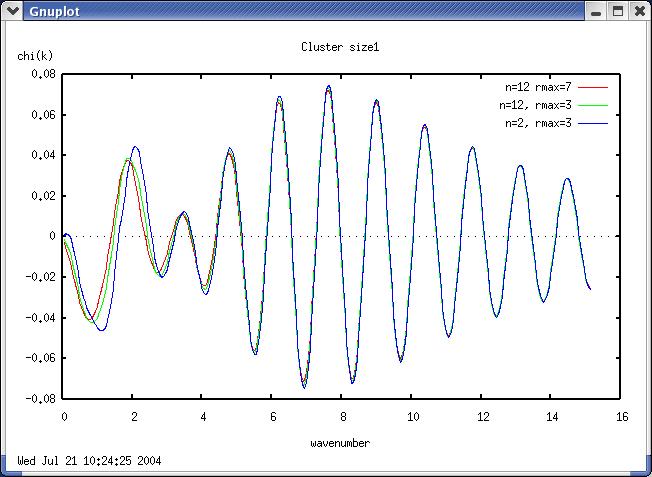
Hi Scott, Feff uses the placement of the atoms to determine the size of the muffin tin radius and the norman radius, which define where the atom is verse where the atom is not. If there are no neighboring atom then these radii become unphysically large and then the scattering amplitudes will not be accurate. -Copper is kinda special. Copper has 12 neighboring atoms, so the muffin tin radii are pretty well constrained even with one shell. If you run a feff calculation with all the Cu atoms in a cluster to 7 Angstroms the top of the feff0001.dat file shows the radii for the atoms and it looks something like this: For large cluster Rmax=7.0 Angstroms: #Copper, Kittel, ISSP Feff 7.02 # metal, fcc # Abs Z=29 Rmt= 1.305 Rnm= 1.454 K shell # Pot 1 Z=29 Rmt= 1.273 Rnm= 1.391 # Gam_ch=1.761E+00 H-L exch # Mu=-5.472E+00 kf=1.724E+00 Vint=-1.679E+01 Rs_int= 2.104 Now do it again for N=12 and Rmax=3.0 For small cluster Rmax=3.0 Angstroms, N=12 # Copper, Kittel, ISSP Feff 7.02 # metal, fcc # Abs Z=29 Rmt= 1.233 Rnm= 1.467 K shell # Pot 1 Z=29 Rmt= 1.282 Rnm= 1.570 # Gam_ch=1.761E+00 H-L exch # Mu=-4.594E+00 kf=1.474E+00 Vint=-1.287E+01 Rs_int= 2.461 And again for N=2 and Rmax =3.0 For small cluster Rmax=3.0 Angstroms, N=2 #Copper, Kittel, ISSP Feff 7.02 # metal, fcc # Abs Z=29 Rmt= 1.256 Rnm= 1.815 K shell # Pot 1 Z=29 Rmt= 1.296 Rnm= 1.872 # Gam_ch=1.761E+00 H-L exch # Mu=-3.892E+00 kf=1.204E+00 Vint=-9.411E+00 Rs_int= 3.013 You can see that the norman radius gets really big for the last case, 1.391 to 1.570 to 1.872 and as a result the scattering amplitude changes. I show a comparison of the chi(k)xk for each of these inputs with a N=1.0, DE=0, SS=0, S02=1.0, DR=0 in the attached clustersize.jpg file. Shelly
-----Original Message----- From: Scott Calvin [mailto:SCalvin@slc.edu] Sent: Wednesday, July 21, 2004 9:53 AM To: XAFS Analysis using Ifeffit Subject: RE: [Ifeffit] Re: Artemis Question
Shelly said:
Feff is truly amazing.
Amen!
1) Feff needs to have a list of atoms that extends well beyond the paths that are actually used in the fit. Always pad the feff.inp file by at least an Angstrom. If you are using feff calculation for the first time. Pad the feff list by one angstrom and then by two angstroms and check that the paths that you are going to use are the same. You do not want to use paths from the edges of the cluster that you give to feff in the feff.inp file. -Hence you need to use a structure like a metal-oxide to model a hydrated metal.
Under what circumstances does FEFF actually need these extra atoms? When I take the copper example included by Artemis, for example, I can rerun the FEFF calculation with a cluster size of 3 angstroms, so that FEFF only has the nearest-neighbors to deal with. The path generated is identical to when a cluster size of 7 angstroms. Certainly if someone is using FEFF8 with a self-consistent field calculation it is necessary to have the cluster extend beyond the furthers paths, but with FEFF6L is this true?
(Incidentally, for the novices out there, I do NOT recommend being satisfied with a single-shell fit when you have a reasonable guess as to the structure beyond the first-shell. One reason can be seen easily with Artemis: paths beyond the first-shell often have low-R "tails" that contribute to the signal even at the first-shell peak. Although the FEFF calculation for the first path may be the same whether or not you also calculated paths further out, the Ifeffit fit will be different depending on whether you include those "outer" paths or not.)
--Scott Calvin Sarah Lawrence College
_______________________________________________ Ifeffit mailing list Ifeffit@millenia.cars.aps.anl.gov http://millenia.cars.aps.anl.gov/mailman/listi> nfo/ifeffit
{kind=link}