
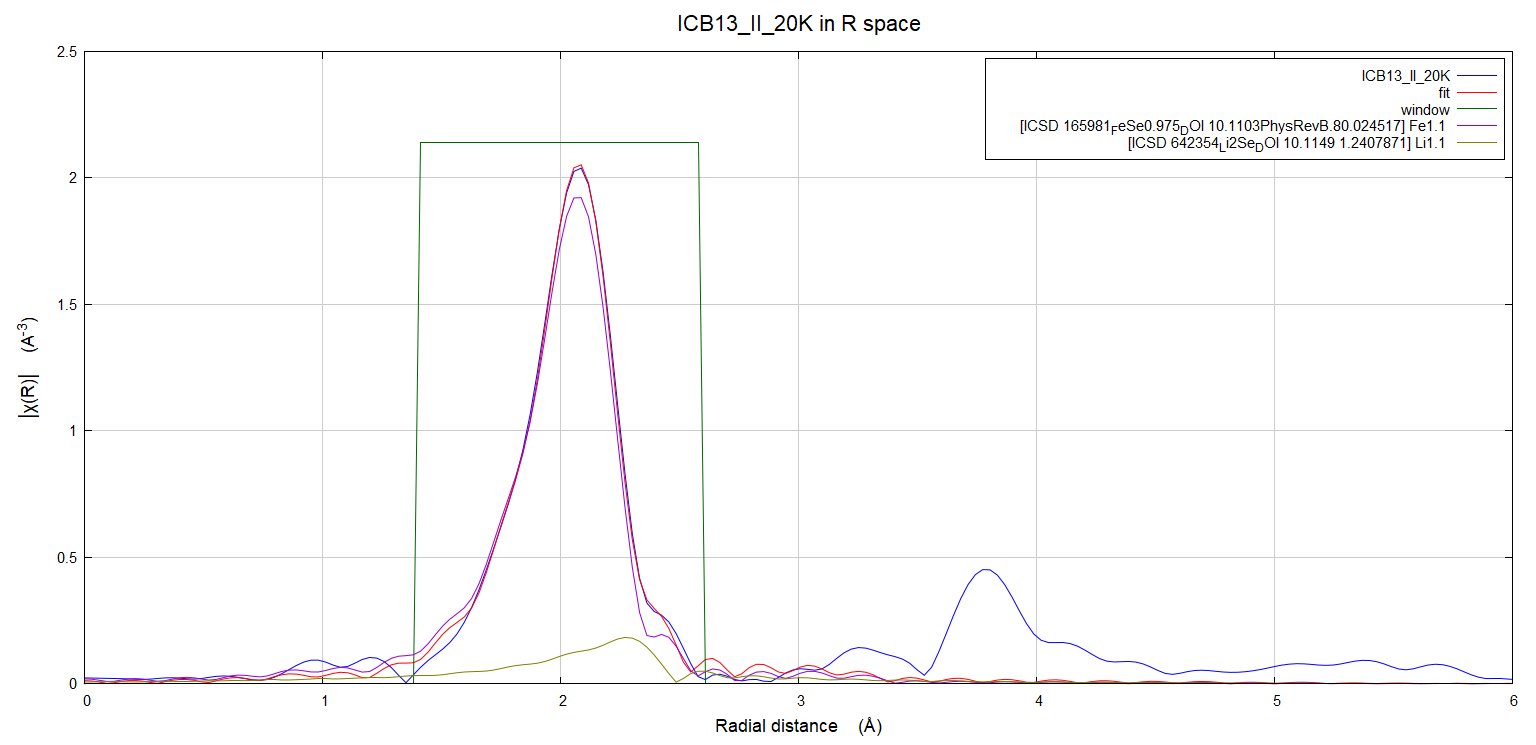
Dear mailing list, I am currently analyzing some EXAFS data. I am studying a Lix(C5H5N)FeSe system in a temperature grid that extends from 20 K to 300 K and I have 4 such datasets which correspond to different amount of doping (x). Right now, im focusing on fitting the 1st coordination cell, in Artemis for the Se K-edge. My starting model is the simple P4/nmm FeSe. So, in my system the 1st coordination cell, in the Se K-edge, corresponds to the Se (absorber) - Fe (backscatterer) pair. I have 2 questions: 1) I realize now, that I have a certain impurity in the high doping range on my system, namely Li2Se. I try to include a scattering path from the respective Li2Se crystal model in my fits, since a Se (absorber) - Li (backscatterer) pair is present in the R-range of my fit in the Forward Fourier Transform. My question here is if this makes sense since Li is much smaller scatterer compared to Se. In other words, does it make sense to look for physical parameters (Li-Se bond length and DW factor respectively) of a signal (Se-Li) that is "tucked" in below the main peak coming from the "majority" Se-Fe signal in the FFT? 2)Also, I'm attempting to extract an Einstein temperature for each of those datasets, by utilizing the "eins(T, thetae)" function implemented in Artemis. What is the equation that is parametrized here? Does it include the s0^2 offset term that accounts for the overall configuration disorder in the system? And if that is the case is there same way to separate it from the temperature dependent s^2 term? Thank you in advance, Deltsidis Alexandros PS:I am attaching a png. file exported from Artemis that is relative to my question 1) Ph.D candidate, Institute of Electronic Structure and Laser (IESL), Foundation for Research and Technology - Hellas (FORTH)
{kind=link}